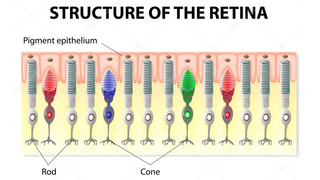
Often diagnosed in childhood or adolescence, Usher syndrome is an inherited disease causing combined hearing loss and vision loss from retinitis pigmentosa. The condition can also cause problems with balance.
Latest News
-
BlueRock Therapeutics and Foundation Fighting Blindness announce collaboration to expand the Uni-Rare natural history study of patients living with inherited retinal diseases
Press ReleasesCollaboration will add a new multi-gene cohort of patients living with inherited retinal diseases. Data insights from the new study cohort will inform the future clinical trial design for BlueRock’s pipeline of cell therapies for treating blindness.
-
Eye on the Cure Podcast – Episode 40: Rebecca Alexander
Foundation PodcastsFebruary 10, 2023. Rebecca Alexander talks candidly with host Ben Shaberman about her journey with Usher syndrome. Reflecting on her own experiences, she discusses how people with vision loss and their loved ones can practice good emotional care and advocate for their needs.
-
Foundation Launching its Largest Natural History Study to Date for 1,500 People with Inherited Retinal Diseases Caused by Rare Mutated Genes
Press ReleasesUni-Rare Study will improve clinical understanding of more IRDs and boost development of potential therapies.
-
Foundation Fighting Blindness and Usher 1F Collaborative to Launch Natural History Study
Press ReleasesThe project will help identify outcome measures for future clinical trials of potential USH1F therapies.
-
Foundation Fighting Blindness To Jointly Host Online Continuing Medical Education (CME, COPE) Webinar
Press ReleasesThe event will review best practices for care and management of patients with inherited retinal diseases such as retinitis pigmentosa, Usher syndrome, and Stargardt disease.
-
Foundation Fighting Blindness Commits $6.5 Million for New Retinal Disease Research Grants
Press ReleasesNew grants include development of CRISPR/Cas9 gene-editing treatments, new disease models, and a retinal regeneration therapy
-
Foundation Insights Forum – July 30, 2020
Insights ForumThe Foundation Fighting Blindness is pleased to provide an audio recording and full transcript of the Insights Forum, our quarterly conference call providing updates to the inherited retinal disease community. The call took place on July 30, 2020.
-
COVID-19 Resources
Foundation NewsThe Foundation Fighting Blindness is closely monitoring the COVID-19 situation and its impact on the IRD community.
-
Foundation Insights Forum – January 31, 2020
Insights ForumThe Foundation Fighting Blindness is pleased to provide an audio recording and full transcripts of the Insights Forum, our quarterly conference call providing updates to the inherited retinal disease community. The call took place on January 31, 2020.
-
ProQR Therapeutics Teams Up with the Foundation Fighting Blindness and Blueprint Genetics to Support the My Retina Tracker® Program for People Living with Inherited Retinal Diseases
Press ReleasesMy Retina Tracker Program is the highest volume IRD genetic testing program in the U.S.
-
Foundation Insights Forum – October 30, 2019
Insights ForumThe Foundation Fighting Blindness is pleased to provide an audio recording and full transcript of the Insights Forum, our quarterly conference call providing updates to the inherited retinal disease community. The call took place on October 30, 2019.
-
Blueprint Genetics, InformedDNA and the Foundation Fighting Blindness launch an open access program for patients with inherited retinal disease in the United States
Press ReleasesThe program will offer patients with inherited retinal disease no-cost genetic testing and genetic counseling in the United States. Look for updated information on how to participate to be posted in mid-October, with program registration starting shortly thereafter.
-
Foundation Fighting Blindness Investing Nearly $6.5 Million in New Grants
Foundation NewsThe newly funded research efforts include several therapies that have strong potential to treat a wide range of inherited retinal diseases.
-
Foundation Fighting Blindness Endorses 'Eye Bonds' Legislation
Press ReleasesBipartisan Bill Will Stimulate Up to $1 Billion in New Funding for Blindness Research
-
Foundation Fighting Blindness Urges Congress to Pass ‘Eye-Bonds’ Legislation
Press ReleasesBill Introduced in U.S. House Would Speed Up Cures for Blindness
-
Foundation Fighting Blindness and CheckedUp® Partner to Educate Retinal-Disease Patients About Research, Resources, and Emerging Therapies During Doctor Visits
Press ReleasesThe Foundation Fighting Blindness (the Foundation) and CheckedUp have formed a collaborative partnership to deliver patient-friendly diagnostic and disease-management information to people with retinal diseases such as age-related macular degeneration, retinitis pigmentosa, and Stargardt disease during their visits to eye doctors.

.png)







Latest Research
-
Usher Syndrome Research Advances
Retinal Disease Research AdvancesRecent developments in research on Usher Syndrome.
-
Théa Completes Acquisition of ProQR’s LCA10 and USH2A Treatment Programs, Plans to Continue Clinical Development
Eye On the Cure Research NewsKnown as antisense oligonucleotides, the treatments performed encouragingly in ProQR’s clinical trials
-
ProQR Seeking to Partner Ophthalmic Programs
Eye On the Cure Research NewsThe company is halting its clinical programs for LCA10 and USH2A as it seeks a new partner
-
Foundation’s RD Fund Invests in SalioGen Therapeutics, Developer of Novel Gene Coding Technology for Treating Inherited Retinal Diseases
Eye On the Cure Research NewsThe company is currently developing programs for Stargardt disease (ABCA4), Usher syndrome, RP25 (EYS), and RP1.
-
ProQR Doses First Patients in Phase 2/3 Clinical Trials for its USH2A-Exon 13 RNA Therapy
Eye On the Cure Research NewsThe Sirius trial is for USH2A (exon 13 mutations) patients with advanced vision loss. The Celeste trial is for USH2A (exon 13 mutations) patients with moderate to early vision loss.
-
Foundation Hosts Workshop on USH1B Research and Therapy Development
Eye On the Cure Research NewsEmerging dual-vector gene therapies to address current cargo-size limitations were highlighted during the meeting.
-
Optogenetics: Hope for Vision Restoration for Advanced Retinal Diseases
Eye On the Cure Research NewsEarly, encouraging results from two human studies — trials launched by Bionic Sight and GenSight — are putting optogenetic therapies in the spotlight for patients with advanced vision loss from retinal conditions.
-
Investigators Report Partial Vision Restoration for One Patient in Optogenetic Therapy Trial
Eye On the Cure Research NewsThe gene-agnostic approach is designed to restore some vision to people with advanced vision loss
-
Foundation Invests $5.5 Million in Seven New Translational Research Projects
Eye On the Cure Research NewsProjects target a variety of conditions including: age-related macular degeneration, Stargardt disease, retinitis pigmentosa, and Usher syndrome type 3A
-
ProQR’s RNA Therapy for USH2A Performs Well in Phase 1/2 Clinical Trial
Eye On the Cure Research NewsThe company is planning two Phase 2/3 clinical trials for the treatment
-
Foundation Invests $3 million in Atsena Therapeutics, New Company Developing GUCY2D-LCA1 and MYO7A-USH1B Gene Therapies
Eye On the Cure Research NewsWith a founding investment from Hatteras Venture Partners, Atsena has raised a total of $8.15 million for its launch.
-
Nacuity’s Emerging Anti-Oxidative Therapy Moves into Clinical Trial
Eye On the Cure Research NewsThe oral treatment shows promise for slowing vision loss in people with RP and Usher syndrome, regardless of genetic profile
-
#GivingTuesdayNow Featured Researcher Dr. Shannon Boye
Eye On the Cure Research NewsA Lifelong Science Nerd is Winning the Fight Against Blindness
-
Interim Results Released for USH2A RNA Therapy Clinical Trial
Eye On the Cure Research NewsSuggestions of efficacy observed in 25 percent of participants receiving the treatment
-
Genetic Testing for Inherited Retinal Diseases through the Foundation’s Open Access Program
Science EducationThe benefits of genetic testing for IRD patients, how to participate in the Foundation’s Open Access program, and what to expect from the genetic testing process.
-
The Retina is a Proving Ground for a Broad Range of Neurological Therapies
Science EducationRetinal research paves the way for new treatments for the entire neurological system.
-
Dr. Don Zack Honored for Research Contributions by ARVO and the Foundation Fighting Blindness
Eye On the Cure Research NewsDr. Zack is a member of the Foundation’s Scientific Advisory Board and chairs its Cellular Molecular Mechanisms of Disease study section.
-
Eye Bonds Re-Introduced to New Congress: Potentially $1 Billion in Government-Backed Funding for Eye Research
Eye On the Cure Research NewsEye Bonds provide the opportunity to advance, and accelerate development for, more promising treatments into and through clinical trials and out to the people who need them.
-
ARVO 2019: Emerging Eloxx Molecules Show Promising Results in Usher Models
Eye On the Cure Research NewsEloxx Pharmaceuticals is developing small molecules that permit read-through of point mutations that cause Usher syndrome 1F and 2A.
-
First Patient Receives ProQR’s Emerging USH2A Therapy in Clinical Trial
Eye On the Cure Research NewsProQR, a developer of RNA therapies in the Netherlands, announced that the first clinical-trial participant has received its emerging treatment, which targets retinitis pigmentosa and Usher syndrome caused by mutations in exon 13 of the USH2A gene.
-
Encouraging Vision Improvements Reported in ReNeuron's Cell-Therapy Clinical Trial
Eye On the Cure Research NewsReNeuron, a cellular therapy developer in the UK, has reported vision improvements in the treated eyes of the first three retinitis pigmentosa (RP) patients in the Phase II part of the Phase I/II clinical trial for its proprietary human retinal progenitor cells (hRPC). The Phase I portion of the trial, completed last year, primarily assessed safety in subjects with minimal remaining vision.
-
The Foundation Receives a $100,000 Research Grant from Sofia Sees Hope
Eye On the Cure Research NewsSofia Sees Hope, a nonprofit dedicated to finding treatments and cures for people with Leber congenital amaurosis (LCA) and other inherited retinal diseases (IRDs), has made a $100,000 donation to the Foundation Fighting Blindness to support therapy development and genetic testing.
-
Pixium's PRIMA Bionic Vision System Restores Central Vision in Dry AMD Clinical Trial
Eye On the Cure Research NewsThe French bioelectronics company Pixium Vision has reported that its PRIMA bionic vision system has restored some central vision in patients with advanced dry age-related macular degeneration (AMD) participating in a clinical feasibility trial.
-
ProQR Receives FDA Authorization to Launch Clinical Trial for USH2A Therapy
Eye On the Cure Research NewsProQR, a biotech in the Netherlands developing therapies for rare diseases, has received authorization from the US Food and Drug Administration to launch a Phase I/II clinical trial for QR-421a, its treatment targeting mutations in exon 13 of the USH2A gene.
-
Foundation Invests $2.5 Million in Search for Elusive Retinal Disease Genes and Mutations
Eye On the Cure Research NewsSince 1989 genetic researchers, many funded by the Foundation, have identified approximately 270 genes linked to IRDs. In most cases, defects in a single gene can cause a retinal disease and vision loss.
-
FFB Congratulates RPE65 Gene Therapy Researchers for Champalimaud Award
Eye On the Cure Research NewsOn September 4, 2018, seven researchers, including six previously funded by the Foundation, were recognized with the prestigious 2018 Antonio Champalimaud Vision Award for their contributions to the advancement of blindness-reversing RPE65 gene therapies.
-
FFB Provides Four Career Development Awards to Up-and-Coming Clinical Researchers
Eye On the Cure Research NewsEach recipient will receive a total of $375,000 over five years to help build an independent research program in addition to their clinical practices.
-
FFB Funding More than $2 Million in New Research
Eye On the Cure Research NewsSeventy scientists submitted requests for funding.
-
Call to Action: Ask Congress to Support $1 Billion in Eye Research
Eye On the Cure Research NewsCall to Action: Ask Congress to Support $1 Billion in Eye Research
-
Retinal Regeneration: Releasing Your Inner Salamander
Eye On the Cure Research NewsMany research groups from around the world are investigating ways to create new photoreceptors from stem cells for transplantation into the retina for vision restoration.
-
ARVO 2018: Dr. Henry Klassen Provides Update on jCyte Stem Cell Trials
Eye On the Cure Research NewsDr. Henry Klassen, jCyte co-founder and investigator at UC Irvine, provides an update on the clinical trials for an RP therapy derived from stem cells.
-
ARVO 2018: Dr. Stephen Daiger Reports on the State of Genetic Testing for Inherited Retinal Diseases
Eye On the Cure Research NewsARVO 2018: Dr. Stephen Daiger Reports on the State of Genetic Testing for Inherited Retinal Diseases
-
ARVO 2018: Dr. Steve Rose Reports on CRISPR/Cas9 for Inherited Retinal Diseases
Eye On the Cure Research NewsFFB’s own Dr. Steve Rose, chief scientific officer, reviews our commitment to funding and exploring CRISPR/Cas9 gene editing for inherited retinal disease.
-
ARVO 2018: World's Largest Show and Tell for Innovations in Eye Research
Eye On the Cure Research NewsMore than 11,000 eye researchers from around the world — including five intrepid members from FFB’s science team — will gather to participate in what is essentially a massive “show and tell” of the latest scientific advancements.
-
Study Suggests Vitamin A May Benefit Children with RP
Eye On the Cure Research NewsAn FFB-funded study at Massachusetts Eye andEar Infirmary (MEEI) suggests that vitamin A palmitate supplementation may slow the decline of cone function by nearly 50 percent in children with retinitis pigmentosa (RP).
-
FFB-CRI Investing $7.5 Million in Emerging Therapy for USH2A
Eye On the Cure Research NewsThe Foundation Fighting Blindness Clinical Research Institute (FFB-CRI) has entered into a partnership with ProQR to develop a retinal therapy for people with Usher syndrome type 2A (USH2A) caused by mutations in exon 13 of the USH2A gene.
-
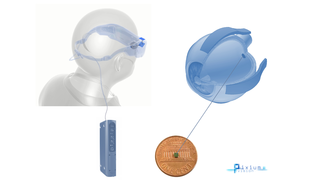
Clinical Trial to Launch for System Combining Optogenetics and Eyewear
Eye On the Cure Research NewsThe French biotech GenSight Biologics has received regulatory authorization in the UK to launch the PIONEER Phase 1 \ 2 clinical trial for its GS030 system — a light-sensing gene therapy (optogenetics) coupled with eyewear, which enhances visual stimulation.
-
Top Retinal Research Advances for 2017
Eye On the Cure Research NewsAn exciting year in fighting blindness.
-
jCyte Reports Results for Phase 1/2a Clinical Trial for Retinal-Cell Treatment
Eye On the Cure Research NewsSome participants reported increased light sensitivity, improved color vision, better mobility, and improved reading ability.
-
History Is Made: FDA Approves Spark's Vision-Restoring Gene Therapy
Eye On the Cure Research NewsKnown as LUXTURNA™ (voretigene neparvovec), the gene therapy restored vision in a clinical trial for people between the ages of 4 and 44 with Leber congenital amaurosis (LCA) caused by mutations in the gene RPE65.
-
Stem-Cell Therapy Clinics Remain Inadequately Regulated, Pose Risk to Patients
Eye On the Cure Research NewsIf a clinic is charging for a stem-cell treatment or procedure for an IRD, it is probably not legit. The expense to the patient is a major red flag.
-
FDA Committee Unanimously Recommends Approval for Spark's RPE65 Gene Therapy - Final Decision Due in January 2018
Eye On the Cure Research NewsAn advisory committee comprised of FDA-selected experts voted unanimously – 16 to 0 – to recommend approval.
-
The Foundation's Investments Are Filling the Pipeline for Vision-Saving Therapies
Eye On the Cure Research NewsIn addition to funding promising biotech start-ups, the Foundation Fighting Blindness has played a critical role in developing research talent.
-
Scientists Receive $25 Million to Develop a Vision-Restoring System that Connects to the Brain
Eye On the Cure Research NewsThe high-tech, vision-restoring system interfaces with the visual cortex, the back of the brain where visual input is processed to create the images we see.
-
Foundation Fighting Blindness and 4D Molecular Therapeutics Partner to Boost Retinal Gene Therapy Development
Eye On the Cure Research NewsThe partnership will help companies and researchers quickly obtain and implement high-quality vectors for their retinal gene-therapy development efforts.
-
FFB-Funded Scientists Report on Nine Promising Translational Research Efforts
Eye On the Cure Research NewsThe Foundation Fighting Blindness has taken the translational challenge head on by investing more than $75 million in therapy-development projects with strong clinical-trial potential through its Translational Research Acceleration Program (TRAP), which includes Gund-Harrington Scholar Awards.
-
SparingVision Formed to Advance Sight-Saving Protein for RP
Eye On the Cure Research NewsSparingVision received a €300,000 award known as the Honor Prize from the French Ministry of Research. The award is given to new, innovative companies in France competing in a national contest.
-
Forty-Four High-Impact Retinal-Research Efforts Highlighted at FFB-Casey Innovation Summit
Eye On the Cure Research NewsIn its fourth year, the meeting is becoming the world’s most comprehensive overview of the promising research underway for emerging IRD treatments.
-
FFB Funding Helps Retinal Genetics Lab Secure $2 Million Investment
Eye On the Cure Research NewsHow the Foundation Fighting Blindness (FFB) provided timely funding of $155,000 to help a lab at the University of California, San Diego (UCSD), leverage a $2 million retinal-gene discovery project.
-
jCyte Stem-Cell Therapy Moves into Phase IIb Clinical Trial for RP
Eye On the Cure Research NewsBased on lab studies, researchers believe the treatment can preserve and potentially rescue the patient’s existing photoreceptors, thereby saving and possibly restoring vision.
-
FFB-CRI Launching Natural History Study for People with USH2A Mutations
Eye On the Cure Research NewsThe study — known as RUSH2A (“R” stands for “rate of progression”) — is beginning in spring 2017 and will take place at about 20 clinical sites around the world. RUSH2A investigators will use a variety of technologies to monitor changes in vision and retinal structure to document and analyze disease progression.
-
Dr. Eliot Berson, Pioneer in Vitamin A Therapy for Retinitis Pigmentosa, Passes Away
Eye On the Cure Research NewsDr. Berson dedicated himself to clinical care and vision-saving research for people with inherited retinal diseases for five decades.
-
Unregulated Stem-Cell Therapy Causes Severe Vision Loss for Three Florida Women
Eye On the Cure Research News“…participation in a study for an emerging therapy that is not regulated by the FDA or another well-recognized regulatory agency like the European Medicines Agency in Europe, is fraught with dangers and can lead to unexpected serious consequences.”
-
AGTC Leverages Funding from the Foundation to Move Promising Treatments into Clinical Trials
Eye On the Cure Research NewsCompany Builds on FFB’s Initial Investment to Garner $265 Million in Therapy Development Funding
-
Foundation Investing in Drug to Slow Many Forms of RP
Eye On the Cure Research NewsThe Foundation Fighting Blindness Clinical Research Institute (FFB-CRI) has announced an investment of up to $7.5 million to advance the potential therapy into and through a Phase II clinical trial.
-
FFB-CRI Leads Effort to Identify Outcome Measures for Therapies in Clinical Trials
Eye On the Cure Research NewsImproved outcome measures will make clinical trials for degenerative retinal diseases — including age-related macular degeneration (AMD), the world’s leading cause of blindness in seniors, and inherited retinal conditions such as RP and Stargardt disease — less expensive to conduct and able to deliver more precise results.
-
A Change in Identity Might Someday Save Vision
Eye On the Cure Research NewsBy changing the identity of cells in the retina–namely rods–researchers may someday be able to slow or halt vision loss for those with retinitis pigmentosa (RP) and other related conditions.
-
Building a Wiring Diagram for the Retina to Help Researchers Save and Restore Vision
Eye On the Cure Research NewsUnderstanding the pathways of the retinal neural network — and how they are rewired with aging and disease — is helpful in trying to save and restore vision.
-
Nobel-Prize-Winning Stem-Cell Researcher Delivers Keynote at FFB-Funded Conference in Kyoto
Eye On the Cure Research NewsDr. Shinya Yamanka discussed his early clinical trial for iPSC-derived retinal pigment epithelial (RPE) cells for a 78-year-old woman with advanced wet age-related macular degeneration (AMD).
-
Embrace Your Exceptions: A Mantra for Understanding Retinal-Disease Inheritance
Eye On the Cure Research NewsThe complex and elusive nature of these conditions can also extend to the way they are passed down in families, making diagnosis and prognosis quite challenging.
-
Optogenetic Therapy Takes First Step Forward in Clinical Trial
Eye On the Cure Research NewsRetroSense’s optogenetic therapy is designed to restore vision to people who are completely blind from retinal degenerative diseases such as retinitis pigmentosa by bestowing light sensitivity to retinal ganglion cells, which survive after photoreceptors, the cells that make vision possible, are lost.
-
Pixium Vision Reports Progress in Development of Two Advanced Bionic Retina Systems
Eye On the Cure Research NewsBoth approaches show strong, near-term potential for providing meaningful vision to people who are otherwise blind from retinal diseases such as retinitis pigmentosa and age-related macular degeneration (AMD).
-
Stem-Cell Therapy for Retinitis Pigmentosa Safe Thus Far in Early Human Study
Eye On the Cure Research NewsThe trial is one of the first-ever for a stem-cell-derived therapy for RP.
-
VISIONS 2016 — Dr. Shomi Bhattacharya Wins FFB Award for Gaining an Understanding of Variations in Vision Loss
Eye On the Cure Research NewsAt VISIONS 2016, FFB’s national conference, the Foundation honored him with its Ed Gollob Board of Directors Award for breakthrough research conducted within the past year.
-
VISIONS 2016 - Dr. Richard Weleber Receives FFB's Highest Research Honor, Recognized in Touching Video
Eye On the Cure Research NewsDr. Weleber became the 10th recipient of the Foundation’s highest honor, named after FFB co-founder Lulie Gund, during the opening lunch of the VISIONS 2016 conference.
-
A Steady Hand in Saving Vision
Eye On the Cure Research NewsSubretinal injection is the most common form of delivery for gene therapies currently in clinical trials.
-
A Leap Forward: Spark Therapeutics Seeks FDA Approval for its Vision-Restoring Gene Therapy
Eye On the Cure Research News -
VISIONS 2015 — Dr. José Sahel Receives Foundation's Most Prestigious Research Honor
Eye On the Cure Research NewsFor those of us supporting the drive for vision-saving treatments and cures, he’s exactly the type of person we want on our team.
-
VISIONS 2015 — Dr. Shannon Boye Receives FFB Award for Excellence in Gene-Therapy Research
Eye On the Cure Research NewsDr. Boye received the Foundation’s Board of Director’s Award, which was presented at VISIONS 2015, FFB’s annual conference, for achievements in retinal research.
-
ARVO 2015 Highlight: The National Eye Institute Invests $4 Million in Audacious-Goals Research
Eye On the Cure Research NewsThe mission of the program—to regenerate the neurons and neural connections in the eye and visual system—is synonymous with the Foundation’s mission to eradicate retinal diseases.
-
ARVO 2015 Highlight: New Research Boosts Prospects for Saving Vision with RdCVF
Eye On the Cure Research NewsAfter years of refinement and testing in animal models, the emerging therapy is about a year and a half from moving into a clinical trial.
-
ARVO 2015 Highlight: A Cut-and-Paste Approach to Fixing Retinal-Disease Genes
Eye On the Cure Research NewsOne of the hot topics at ARVO this year is a rapidly advancing gene-therapy approach called clustered regularly interspaced short palindromic repeats, or CRISPR.
-
Inspired by Progress in Usher Syndrome Research
Eye On the Cure Research NewsMark Pennesi, M.D., Ph.D., a Foundation-funded clinical researcher at the Casey Eye Institute, discusses the first human study of gene therapy for Usher syndrome type 1B.
-
How Evolution is Leading to Gene Therapies for More Retinal Diseases
Eye On the Cure Research NewsAn innovative genetic-engineering approach called “directed evolution” to find optimal gene-delivery systems based on adeno-associated viruses (AAVs).
-
VISIONS 2014 — The Multi-Talented Dr. Shannon Boye
Eye On the Cure Research NewsDr. Boye and her research team received a $900,000 grant for a gene therapy project targeting Leber congenital amaurosis.
-
VISIONS 2014 — My Retina Tracker: Track Your Vision and Drive the Research
Eye On the Cure Research NewsThe powerful and secure system enables patients to keep track of their clinical care and vision changes. At the same time, it enables scientists to search the “de-identified” (i.e., anonymous) patient information to study conditions and identify targets for treatments, preventions and cures.
-
ARVO 2014: European Collaboration Developing Cross-Cutting, Vision-Saving Therapies
Eye On the Cure Research NewsSimply put, they’re creating therapies that can save vision in as many people as possible, independent of the genetic cause of disease.
-
Total Blindness and Non-24 Sleep Disorder
Eye On the Cure Research NewsNon-24 is a very rare condition affecting many (but not all) people who are totally blind and have absolutely no light perception. Their circadian clocks become out of sync as a result.
-
UCI Stem-Cell Pioneer Poised to Launch Clinical Trial for RP Patients
Eye On the Cure Research NewsDr. Henry Klassen’s progenitor-based therapy has the potential to rescue a variety of retinal cells — including rods, cones, retinal pigment epithelium and ganglion cells — and, therefore, may save vision in people with a wide range of conditions.
-
Nouvelle Lumière: French Bionic Retina in a Human Study
Eye On the Cure Research NewsThe French retinal implant developer Pixium quietly launched a clinical trial for its Intelligent Retinal Implant System 1 (IRIS1) in France, Austria and Germany.
-
Is Acupuncture a Beneficial Treatment for Retinitis Pigmentosa?
Eye On the Cure Research NewsAcupuncture definitely has potential benefits, and the breadth of those is being aggressively explored.
-
New Imaging Technique May Be Game-Changer for RP Clinical Trials
Eye On the Cure Research NewsKnown as EZ Width, it holds potential for reducing the time, cost and number of patients needed to determine if a therapy is working in a clinical trial.
-
When a Condition is More than a Retinal Disease
Science EducationThe Foundation Fighting Blindness is, of course, all about finding treatments and cures for retinal degenerative diseases. However, we are well aware that many of our constituents and their families are dealing with more than just vision loss. That’s because genetic defects causing retinal conditions can sometimes affect other parts of the body. The result is conditions often referred to as syndromes.
-
Researchers Move Closer to Getting a Complete Genetic Picture of the Retina
Eye On the Cure Research NewsIdentifying the genes and proteins that play a major role in retinal health and vision is an important step in finding preventions and cures for degenerative diseases.
-
Patient Registries Help Advance Research for Rare Diseases
Eye On the Cure Research NewsMany registries enable patients to collect and track information about their health, so they can take an active role in managing their care.
-
Grow Your Own: Harnessing Muller Glia for Retinal Regeneration
Eye On the Cure Research NewsThere’s hope for retinal regeneration for humans, thanks to Foundation-funded researcher Dr. Thomas Reh, who is investigating how to derive new photoreceptors from retinal cells called Muller glia.
-
Retinal Regeneration is Major Focus of NEI's Audacious Goal
Eye On the Cure Research NewsThe goal, “to regenerate the neurons and neural connections in the eye and visual system,” is exactly what people with retinal diseases need to save and restore their vision.
-
Researcher Revolutionized Fight Against Blindness and Cancer
Eye On the Cure Research NewsA profile on Dr. Robert Langer, a medical researcher who has received dozens of awards, accolades and honorary degrees, including, recently, FFB’s Visionary Award.
-
Staying Alive: Saving Retinal Cells to Preserve Vision
Science EducationSometimes, saving vision simply comes down to keeping retinal cells alive, or at least slowing their degeneration.
-
History in the Making
Eye On the Cure Research NewsMore good news about treatments and technological advances for restoring vision for people with retinal diseases.
-
Have I Got a Cure for You! Debunking an Alleged Treatment on the Internet
Eye On the Cure Research NewsHow do you know if a treatment is legit? There should be preclinical and clinical trial data published in a peer-reviewed journal on research for the treatment.






















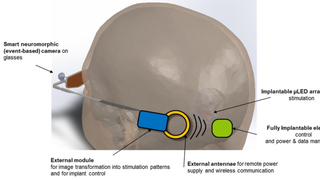







.png)











Related Resources
-
IRD Community Loses Usher Syndrome Research Trailblazer William Kimberling, PhD
Beacon StoriesDr. Kimberling helped discover the identification and characterization of several genes that cause Usher syndrome.
-
Blind Author with Usher Syndrome Ignites the Literary World
Beacon StoriesAt the age of 22, Michael Garrigan received a life-changing diagnosis of Usher syndrome type 2. His vision gradually deteriorated over the years, and by the age of 45, he faced the reality of being legally blind. Determined to share his story and shed light on the capacity to triumph over darkness, Michael penned a remarkable memoir, “Ushered Out of Darkness,” in which he invites readers into his most vulnerable life moments and victories.
-
Breaking Barriers as an Attorney With Usher Syndrome
Beacon StoriesTara is a woman of extraordinary talents. A beloved twin sister, mother of two, and wife of 10 years, Tara leads a fulfilling life dedicated to advocating for others through law.
-
Rewriting the Narrative as the First Black Deaf-Blind Journalist
Beacon StoriesSteven McCoy has made history as the first black deaf-blind journalist in the world, as he was recently diagnosed with Usher syndrome after a lifelong struggle with hearing loss. Equipped with a passion for storytelling and advocacy, he now uses his voice to create lasting change within the entertainment industry and deaf-blind community.
-
Birding Blind: Identifying Birds by Song
Beacon StoriesMartha Steele has been birding for over 30 years, but the way in which she birds has changed over time. Martha has Usher syndrome, with her central vision declining rapidly in the early 2000s and receiving her first cochlear implant in 2010. Now she birds entirely by ear and has learned the songs of about 150 bird species.
-
Biologist and Father Dedicated to Daughter’s Cure
Beacon StoriesAfter Allison’s diagnosis with Usher syndrome type 3, her dad, Jeff Libby, wanted to do everything he could to find her a cure. As a biologist, Jeff started searching for organizations researching blinding diseases like Allison’s, and he found the Foundation Fighting Blindness.
-
Advocating for the Foundation Through Professional Outreach
Beacon StoriesAs a longtime supporter and advocate for the Foundation Fighting Blindness, Lora has recently begun working with the Professional Outreach team. Lora’s work helps eye care professionals in the Philadelphia region provide vital resources for their patients with inherited retinal diseases.
-
Girl Scouts are Changing the World One Crosswalk at a Time
Beacon StoriesInspired by their troop leader diagnosed with Usher syndrome, Girl Scout Troop 1673 is hoping to change the world, one crosswalk at a time, with their Paint It Yellow challenge.
-
Ambitious Swimmer Spreading Optimism
Beacon Stories26-year-old Becca is a two-time Paralympic swimmer for Team USA, diagnosed with Usher syndrome type 1 (USH1) at only four years old. But she has never let her diagnosis with Usher syndrome stop her from doing exactly what she puts her mind to.
-
ProQR Announces Virtual Presentations at Scientific Conferences
ResourceProQR Therapeutics N.V. (Nasdaq:PRQR), a company dedicated to changing lives through the creation of transformative RNA therapies for severe genetic rare diseases, today announced virtual presentations at the Ophthalmology Futures Retina Forum, European Society of Retina Specialists (Euretina) congress and the Annual Meeting of the American Academy of Optometry (AAOpt).
-
An Artist, First and Foremost
Beacon StoriesAllen has always wanted to be known as an artist, first and foremost. His photography hints at the ever-changing nature of people’s lives and their environment, much like his own progression with retinitis pigmentosa (RP).
-
Blind Miracle on Ice
Beacon StoriesShawn was diagnosed with Usher syndrome at a young age. Shawn now stays involved with the blind community and participates in his local blind hockey league. In his own words, he shares his journey at the Toyota-USA Hockey Disabled Hockey Festival.
-
Artist with Usher Syndrome Excited to Register on My Retina Tracker to Drive Retinal Research
Beacon StoriesArtist Dana Simon describes her experience with My Retina Tracker, a free and secure online registry for people with inherited retinal diseases.
-
Ready for the Spotlight: Rebecca Alexander Shares Her Story of Living with Usher Syndrome
Beacon Stories“…the best thing is to reach out and network, and access all the resources. The more invested you are in what’s out there, the more you get back.”
-
Flight4Sight: A Man with Usher Syndrome Travels Around the Globe
Beacon StoriesThere’s nothing like a sense of urgency to turn what seems like a crazy idea into reality.
-
An FFB Board Member’s Perspective on Her Experience with Acupuncture
Beacon StoriesMoira Shea describes her experience with acupuncture.
-
Reflections on Life with Usher Syndrome
Beacon StoriesFoundation Board Member Moira Shea describes her life with a retinal disease.




















