
X-Linked Retinoschisis (XLRS)
X-linked retinoschisis (XLRS) is an inherited retinal disease causing vision loss due to splitting of the layers of the retina.
What is X-Linked Retinoschisis?
X-linked retinoschisis (XLRS) is an inherited disease that causes loss of central and peripheral vision due to degeneration of the retina. The retina is a thin piece of tissue lining the back of the eye. Rod and cone photoreceptors in the retina convert light into electrical signals that the brain interprets as vision. About 35,000 people in the United States and Europe have the condition.
XLRS leads to vision loss due to the splitting of retinal layers, which leads to degeneration of photoreceptors. It is caused by mutations in the RS1 gene. The RS1 protein act likes an adhesive to hold the retinal layers together.
Symptoms
As an X-linked disease, XLRS occurs primarily in males. The condition is typically diagnosed in childhood. One of the first signs of XLRS is loss of visual acuity that is not correctable. Vision loss is often progressive leading to legal blindness.
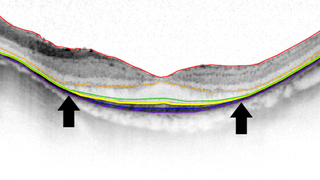
Cystic macular lesions (like blisters) in the retina are the hallmark feature of XLRS. While these lesions can increase retinal thickness thereby exacerbating vision loss, they can be treated with medications including oral or topical carbonic anhydrase inhibitors.
The most common gene associated with X-linked retinoschisis is RS1.
Inheritance
As an X-linked disease, the mutated gene, RS1, is located on the X chromosome. Females have two X chromosomes and can carry the disease gene on one of their X chromosomes. Because they have a healthy version of the gene on their other X chromosome, carrier females are less frequently affected by X-linked diseases. Males have only one X chromosome (paired with one Y chromosome) and are therefore genetically susceptible to X-linked diseases. Males with X-linked diseases pass their Y chromosome to their sons, and therefore will never pass an X-linked disease to their sons. Female carriers have a 50 percent chance (or 1 chance in 2) of passing the X-linked disease gene to their daughters, who become carriers, and a 50 percent chance of passing the gene to their sons, who are then affected by the disease.
Female carriers have a 50 percent chance (or 1 chance in 2) of passing the X-linked disease gene to their daughters, who become carriers, and a 50 percent chance of passing the gene to their sons, who are then affected by the disease.
Genetic counselors are excellent resources for discussing inheritability, family planning, genetic testing, and other related issues.
Living with the Disease
There are many services and accommodative and assistive resources available to people and families with X-linked retinoschisis. Visit the Foundation’s Low Vision Resources page to learn about many of these resources. A low vision specialist can help recommend the resources that are right for you.
More information on living with XLRS is in the Newly Diagnosed section of this Web site.
Genetic Testing
Genetic testing is available for XLRS. It helps with attaining an accurate diagnosis and assessing the risk of passing the disorder from parent to offspring. A patient with an accurate diagnosis is in a better position to understand which emerging treatment approaches and clinical trials are most appropriate for them.
Clinical Trials
View a list of current clinical trials, many made possible by Foundation support
XLRS page at www.ClinicalTrials.gov.
Resource Download
Download What You Should Know about X-Linked Retinoschisis Infographic - 11x17
Download What You Should Know about X-Linked Retinoschisis Infographic - 11x17 (PNG, 37.3 KB)
Next Section
Read the Most Recent Research on X-Linked Retinoschisis (XLRS)
Latest News
-
COVID-19 Resources
Foundation NewsThe Foundation Fighting Blindness is closely monitoring the COVID-19 situation and its impact on the IRD community.
-
Foundation Insights Forum – January 31, 2020
Insights ForumThe Foundation Fighting Blindness is pleased to provide an audio recording and full transcripts of the Insights Forum, our quarterly conference call providing updates to the inherited retinal disease community. The call took place on January 31, 2020.
-
ProQR Therapeutics Teams Up with the Foundation Fighting Blindness and Blueprint Genetics to Support the My Retina Tracker® Program for People Living with Inherited Retinal Diseases
Press ReleasesMy Retina Tracker Program is the highest volume IRD genetic testing program in the U.S.
-
Foundation Insights Forum – October 30, 2019
Insights ForumThe Foundation Fighting Blindness is pleased to provide an audio recording and full transcript of the Insights Forum, our quarterly conference call providing updates to the inherited retinal disease community. The call took place on October 30, 2019.
-
Blueprint Genetics, InformedDNA and the Foundation Fighting Blindness launch an open access program for patients with inherited retinal disease in the United States
Press ReleasesThe program will offer patients with inherited retinal disease no-cost genetic testing and genetic counseling in the United States. Look for updated information on how to participate to be posted in mid-October, with program registration starting shortly thereafter.
-
Foundation Fighting Blindness Investing Nearly $6.5 Million in New Grants
Foundation NewsThe newly funded research efforts include several therapies that have strong potential to treat a wide range of inherited retinal diseases.
-
Foundation Fighting Blindness Endorses 'Eye Bonds' Legislation
Press ReleasesBipartisan Bill Will Stimulate Up to $1 Billion in New Funding for Blindness Research
-
Foundation Fighting Blindness Urges Congress to Pass ‘Eye-Bonds’ Legislation
Press ReleasesBill Introduced in U.S. House Would Speed Up Cures for Blindness
-
Foundation Fighting Blindness and CheckedUp® Partner to Educate Retinal-Disease Patients About Research, Resources, and Emerging Therapies During Doctor Visits
Press ReleasesThe Foundation Fighting Blindness (the Foundation) and CheckedUp have formed a collaborative partnership to deliver patient-friendly diagnostic and disease-management information to people with retinal diseases such as age-related macular degeneration, retinitis pigmentosa, and Stargardt disease during their visits to eye doctors.




Latest Research
-
Atsena Doses First Patient in XLRS Gene Therapy Clinical Trial
Eye On the Cure Research NewsThe AAV.SPR gene delivery system used in the trial is designed to more safely reach targeted retinal cells
-
Atsena Receives FDA Authorization to Launch a Gene Therapy Clinical Trial for XLRS
Eye On the Cure Research NewsKnown as The Lighthouse Study, the Phase ½ trial is expected to begin in mid-2023
-
Atsena Therapeutics Developing X-Linked Retinoschisis Gene Therapy
Eye On the Cure Research NewsThe emerging gene therapy is being designed to more safely reach the fovea
-
MeiraGTx and Janssen Pharmaceuticals Report Promising Interim Results from its Phase 1/2 Clinical Trial for XLRP Gene Therapy
Eye On the Cure Research NewsA Phase 3 trial for the treatment is planned
-
Genetic Testing for Inherited Retinal Diseases through the Foundation’s Open Access Program
Science EducationThe benefits of genetic testing for IRD patients, how to participate in the Foundation’s Open Access program, and what to expect from the genetic testing process.
-
The Retina is a Proving Ground for a Broad Range of Neurological Therapies
Science EducationRetinal research paves the way for new treatments for the entire neurological system.
-
Dr. Don Zack Honored for Research Contributions by ARVO and the Foundation Fighting Blindness
Eye On the Cure Research NewsDr. Zack is a member of the Foundation’s Scientific Advisory Board and chairs its Cellular Molecular Mechanisms of Disease study section.
-
Eye Bonds Re-Introduced to New Congress: Potentially $1 Billion in Government-Backed Funding for Eye Research
Eye On the Cure Research NewsEye Bonds provide the opportunity to advance, and accelerate development for, more promising treatments into and through clinical trials and out to the people who need them.
-
The Foundation Receives a $100,000 Research Grant from Sofia Sees Hope
Eye On the Cure Research NewsSofia Sees Hope, a nonprofit dedicated to finding treatments and cures for people with Leber congenital amaurosis (LCA) and other inherited retinal diseases (IRDs), has made a $100,000 donation to the Foundation Fighting Blindness to support therapy development and genetic testing.
-
Pixium's PRIMA Bionic Vision System Restores Central Vision in Dry AMD Clinical Trial
Eye On the Cure Research NewsThe French bioelectronics company Pixium Vision has reported that its PRIMA bionic vision system has restored some central vision in patients with advanced dry age-related macular degeneration (AMD) participating in a clinical feasibility trial.
-
Foundation Invests $2.5 Million in Search for Elusive Retinal Disease Genes and Mutations
Eye On the Cure Research NewsSince 1989 genetic researchers, many funded by the Foundation, have identified approximately 270 genes linked to IRDs. In most cases, defects in a single gene can cause a retinal disease and vision loss.
-
FFB Congratulates RPE65 Gene Therapy Researchers for Champalimaud Award
Eye On the Cure Research NewsOn September 4, 2018, seven researchers, including six previously funded by the Foundation, were recognized with the prestigious 2018 Antonio Champalimaud Vision Award for their contributions to the advancement of blindness-reversing RPE65 gene therapies.
-
FFB Provides Four Career Development Awards to Up-and-Coming Clinical Researchers
Eye On the Cure Research NewsEach recipient will receive a total of $375,000 over five years to help build an independent research program in addition to their clinical practices.
-
FFB Funding More than $2 Million in New Research
Eye On the Cure Research NewsSeventy scientists submitted requests for funding.
-
Call to Action: Ask Congress to Support $1 Billion in Eye Research
Eye On the Cure Research NewsCall to Action: Ask Congress to Support $1 Billion in Eye Research
-
Retinal Regeneration: Releasing Your Inner Salamander
Eye On the Cure Research NewsMany research groups from around the world are investigating ways to create new photoreceptors from stem cells for transplantation into the retina for vision restoration.
-
ARVO 2018: Dr. Stephen Daiger Reports on the State of Genetic Testing for Inherited Retinal Diseases
Eye On the Cure Research NewsARVO 2018: Dr. Stephen Daiger Reports on the State of Genetic Testing for Inherited Retinal Diseases
-
ARVO 2018: Dr. Steve Rose Reports on CRISPR/Cas9 for Inherited Retinal Diseases
Eye On the Cure Research NewsFFB’s own Dr. Steve Rose, chief scientific officer, reviews our commitment to funding and exploring CRISPR/Cas9 gene editing for inherited retinal disease.
-
ARVO 2018: World's Largest Show and Tell for Innovations in Eye Research
Eye On the Cure Research NewsMore than 11,000 eye researchers from around the world — including five intrepid members from FFB’s science team — will gather to participate in what is essentially a massive “show and tell” of the latest scientific advancements.
-
Clinical Trial to Launch for System Combining Optogenetics and Eyewear
Eye On the Cure Research NewsThe French biotech GenSight Biologics has received regulatory authorization in the UK to launch the PIONEER Phase 1 \ 2 clinical trial for its GS030 system — a light-sensing gene therapy (optogenetics) coupled with eyewear, which enhances visual stimulation.
-
Top Retinal Research Advances for 2017
Eye On the Cure Research NewsAn exciting year in fighting blindness.
-
History Is Made: FDA Approves Spark's Vision-Restoring Gene Therapy
Eye On the Cure Research NewsKnown as LUXTURNA™ (voretigene neparvovec), the gene therapy restored vision in a clinical trial for people between the ages of 4 and 44 with Leber congenital amaurosis (LCA) caused by mutations in the gene RPE65.
-
Stem-Cell Therapy Clinics Remain Inadequately Regulated, Pose Risk to Patients
Eye On the Cure Research NewsIf a clinic is charging for a stem-cell treatment or procedure for an IRD, it is probably not legit. The expense to the patient is a major red flag.
-
FDA Committee Unanimously Recommends Approval for Spark's RPE65 Gene Therapy - Final Decision Due in January 2018
Eye On the Cure Research NewsAn advisory committee comprised of FDA-selected experts voted unanimously – 16 to 0 – to recommend approval.
-
The Foundation's Investments Are Filling the Pipeline for Vision-Saving Therapies
Eye On the Cure Research NewsIn addition to funding promising biotech start-ups, the Foundation Fighting Blindness has played a critical role in developing research talent.
-
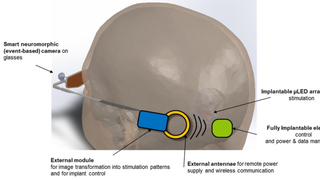
Scientists Receive $25 Million to Develop a Vision-Restoring System that Connects to the Brain
Eye On the Cure Research NewsThe high-tech, vision-restoring system interfaces with the visual cortex, the back of the brain where visual input is processed to create the images we see.
-
Foundation Fighting Blindness and 4D Molecular Therapeutics Partner to Boost Retinal Gene Therapy Development
Eye On the Cure Research NewsThe partnership will help companies and researchers quickly obtain and implement high-quality vectors for their retinal gene-therapy development efforts.
-
FFB-Funded Scientists Report on Nine Promising Translational Research Efforts
Eye On the Cure Research NewsThe Foundation Fighting Blindness has taken the translational challenge head on by investing more than $75 million in therapy-development projects with strong clinical-trial potential through its Translational Research Acceleration Program (TRAP), which includes Gund-Harrington Scholar Awards.
-
Twelve People Receive XLRS Gene Therapy in AGTC's Clinical Trial
Eye On the Cure Research NewsAGTC’s gene therapy uses a human-engineered virus — and adeno-associated virus or AAV — to deliver normal copies of retinoschisin to the patient’s retina.
-
Forty-Four High-Impact Retinal-Research Efforts Highlighted at FFB-Casey Innovation Summit
Eye On the Cure Research NewsIn its fourth year, the meeting is becoming the world’s most comprehensive overview of the promising research underway for emerging IRD treatments.
-
FFB Funding Helps Retinal Genetics Lab Secure $2 Million Investment
Eye On the Cure Research NewsHow the Foundation Fighting Blindness (FFB) provided timely funding of $155,000 to help a lab at the University of California, San Diego (UCSD), leverage a $2 million retinal-gene discovery project.
-
Dr. Eliot Berson, Pioneer in Vitamin A Therapy for Retinitis Pigmentosa, Passes Away
Eye On the Cure Research NewsDr. Berson dedicated himself to clinical care and vision-saving research for people with inherited retinal diseases for five decades.
-
Unregulated Stem-Cell Therapy Causes Severe Vision Loss for Three Florida Women
Eye On the Cure Research News“…participation in a study for an emerging therapy that is not regulated by the FDA or another well-recognized regulatory agency like the European Medicines Agency in Europe, is fraught with dangers and can lead to unexpected serious consequences.”
-
AGTC Leverages Funding from the Foundation to Move Promising Treatments into Clinical Trials
Eye On the Cure Research NewsCompany Builds on FFB’s Initial Investment to Garner $265 Million in Therapy Development Funding
-
FFB-CRI Leads Effort to Identify Outcome Measures for Therapies in Clinical Trials
Eye On the Cure Research NewsImproved outcome measures will make clinical trials for degenerative retinal diseases — including age-related macular degeneration (AMD), the world’s leading cause of blindness in seniors, and inherited retinal conditions such as RP and Stargardt disease — less expensive to conduct and able to deliver more precise results.
-
Building a Wiring Diagram for the Retina to Help Researchers Save and Restore Vision
Eye On the Cure Research NewsUnderstanding the pathways of the retinal neural network — and how they are rewired with aging and disease — is helpful in trying to save and restore vision.
-
Nobel-Prize-Winning Stem-Cell Researcher Delivers Keynote at FFB-Funded Conference in Kyoto
Eye On the Cure Research NewsDr. Shinya Yamanka discussed his early clinical trial for iPSC-derived retinal pigment epithelial (RPE) cells for a 78-year-old woman with advanced wet age-related macular degeneration (AMD).
-
Embrace Your Exceptions: A Mantra for Understanding Retinal-Disease Inheritance
Eye On the Cure Research NewsThe complex and elusive nature of these conditions can also extend to the way they are passed down in families, making diagnosis and prognosis quite challenging.
-
Optogenetic Therapy Takes First Step Forward in Clinical Trial
Eye On the Cure Research NewsRetroSense’s optogenetic therapy is designed to restore vision to people who are completely blind from retinal degenerative diseases such as retinitis pigmentosa by bestowing light sensitivity to retinal ganglion cells, which survive after photoreceptors, the cells that make vision possible, are lost.
-
Pixium Vision Reports Progress in Development of Two Advanced Bionic Retina Systems
Eye On the Cure Research NewsBoth approaches show strong, near-term potential for providing meaningful vision to people who are otherwise blind from retinal diseases such as retinitis pigmentosa and age-related macular degeneration (AMD).
-
VISIONS 2016 — Dr. Shomi Bhattacharya Wins FFB Award for Gaining an Understanding of Variations in Vision Loss
Eye On the Cure Research NewsAt VISIONS 2016, FFB’s national conference, the Foundation honored him with its Ed Gollob Board of Directors Award for breakthrough research conducted within the past year.
-
VISIONS 2016 - Dr. Richard Weleber Receives FFB's Highest Research Honor, Recognized in Touching Video
Eye On the Cure Research NewsDr. Weleber became the 10th recipient of the Foundation’s highest honor, named after FFB co-founder Lulie Gund, during the opening lunch of the VISIONS 2016 conference.
-
A Steady Hand in Saving Vision
Eye On the Cure Research NewsSubretinal injection is the most common form of delivery for gene therapies currently in clinical trials.
-
A Leap Forward: Spark Therapeutics Seeks FDA Approval for its Vision-Restoring Gene Therapy
Eye On the Cure Research News -
VISIONS 2015 — Dr. José Sahel Receives Foundation's Most Prestigious Research Honor
Eye On the Cure Research NewsFor those of us supporting the drive for vision-saving treatments and cures, he’s exactly the type of person we want on our team.
-
VISIONS 2015 — Dr. Shannon Boye Receives FFB Award for Excellence in Gene-Therapy Research
Eye On the Cure Research NewsDr. Boye received the Foundation’s Board of Director’s Award, which was presented at VISIONS 2015, FFB’s annual conference, for achievements in retinal research.
-
ARVO 2015 Highlight: The National Eye Institute Invests $4 Million in Audacious-Goals Research
Eye On the Cure Research NewsThe mission of the program—to regenerate the neurons and neural connections in the eye and visual system—is synonymous with the Foundation’s mission to eradicate retinal diseases.
-
ARVO 2015 Highlight: A Cut-and-Paste Approach to Fixing Retinal-Disease Genes
Eye On the Cure Research NewsOne of the hot topics at ARVO this year is a rapidly advancing gene-therapy approach called clustered regularly interspaced short palindromic repeats, or CRISPR.
-
How Evolution is Leading to Gene Therapies for More Retinal Diseases
Eye On the Cure Research NewsAn innovative genetic-engineering approach called “directed evolution” to find optimal gene-delivery systems based on adeno-associated viruses (AAVs).
-
VISIONS 2014 — My Retina Tracker: Track Your Vision and Drive the Research
Eye On the Cure Research NewsThe powerful and secure system enables patients to keep track of their clinical care and vision changes. At the same time, it enables scientists to search the “de-identified” (i.e., anonymous) patient information to study conditions and identify targets for treatments, preventions and cures.
-
VISIONS 2014 — The Multi-Talented Dr. Shannon Boye
Eye On the Cure Research NewsDr. Boye and her research team received a $900,000 grant for a gene therapy project targeting Leber congenital amaurosis.
-
ARVO 2014: European Collaboration Developing Cross-Cutting, Vision-Saving Therapies
Eye On the Cure Research NewsSimply put, they’re creating therapies that can save vision in as many people as possible, independent of the genetic cause of disease.
-
Total Blindness and Non-24 Sleep Disorder
Eye On the Cure Research NewsNon-24 is a very rare condition affecting many (but not all) people who are totally blind and have absolutely no light perception. Their circadian clocks become out of sync as a result.
-
Nouvelle Lumière: French Bionic Retina in a Human Study
Eye On the Cure Research NewsThe French retinal implant developer Pixium quietly launched a clinical trial for its Intelligent Retinal Implant System 1 (IRIS1) in France, Austria and Germany.
-
Researchers Move Closer to Getting a Complete Genetic Picture of the Retina
Eye On the Cure Research NewsIdentifying the genes and proteins that play a major role in retinal health and vision is an important step in finding preventions and cures for degenerative diseases.
-
Patient Registries Help Advance Research for Rare Diseases
Eye On the Cure Research NewsMany registries enable patients to collect and track information about their health, so they can take an active role in managing their care.
-
Grow Your Own: Harnessing Muller Glia for Retinal Regeneration
Eye On the Cure Research NewsThere’s hope for retinal regeneration for humans, thanks to Foundation-funded researcher Dr. Thomas Reh, who is investigating how to derive new photoreceptors from retinal cells called Muller glia.
-
Retinal Regeneration is Major Focus of NEI's Audacious Goal
Eye On the Cure Research NewsThe goal, “to regenerate the neurons and neural connections in the eye and visual system,” is exactly what people with retinal diseases need to save and restore their vision.
-
Researcher Revolutionized Fight Against Blindness and Cancer
Eye On the Cure Research NewsA profile on Dr. Robert Langer, a medical researcher who has received dozens of awards, accolades and honorary degrees, including, recently, FFB’s Visionary Award.
-
Staying Alive: Saving Retinal Cells to Preserve Vision
Science EducationSometimes, saving vision simply comes down to keeping retinal cells alive, or at least slowing their degeneration.
-
History in the Making
Eye On the Cure Research NewsMore good news about treatments and technological advances for restoring vision for people with retinal diseases.
-
Have I Got a Cure for You! Debunking an Alleged Treatment on the Internet
Eye On the Cure Research NewsHow do you know if a treatment is legit? There should be preclinical and clinical trial data published in a peer-reviewed journal on research for the treatment.


















.png)







Related Resources
-
Mental Health Awareness: Shattering the Stigma
Beacon StoriesAthletes around the world navigate the challenges of daily life and rigorous training, but what if a factor came into play that you had less control over? What if, one day, something changed in your vision?
-
Blind Paralympian Reaching New Heights
Beacon StoriesIsaac Jean-Paul was diagnosed with X-linked juvenile retinoschisis (XLRS) at the age of two. Isaac was always set on being a professional athlete, and now he’s a Paralympian, winning bronze in the long jump in the 2020 Games and breaking the high jump world record three times.
-
Reaching for the Stars
Beacon StoriesAt only three months old, Brendon was diagnosed with x-linked juvenile retinoschisis (XLRS). But with several other family members also with XLRS, Brendon knew he could still follow his passions for science and space. Now 22, Brendon is an Aerospace Engineer, starting a PhD program at UCF, and was recently awarded the prestigious National Science Foundation’s Graduate Research Fellowship.








{kind=link}