
-
SalioGen Developing Novel Gene Insertion Therapy for Stargardt Disease
Eye On the Cure Research News -
Eye on the Cure Podcast | Episode 64: Dr. Kapil Bharti
Foundation Podcasts -
Phase 3 Clinical Trial of NAC Launched for RP Patients
Eye On the Cure Research News

-
ViGeneron Launches Clinical Trial of Gene Therapy for RP Caused by CNGA1 Mutations
Eye On the Cure Research News -
Ocugen to Launch Phase 3 Clinical Trial of Modifier Gene Therapy for RP
Eye On the Cure Research News -
What Does “Blindness” Really Mean?
Beacon Stories

.png)

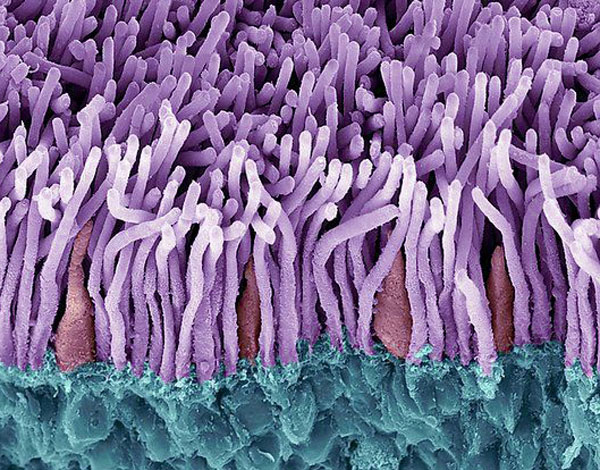
Our Impact
The Foundation is committed to finding treatments and cures for people affected by blinding retinal diseases.
-
1st gene therapy
for any inherited disease to be approved by the FDA
learn more -
$915M+ dollars raised
since 1971
learn more -
35 VisionWalk events
throughout the United States
learn more -
93 grants awarded
in 2023 – over 96 investigators, 71 institutions
Learn more
The Foundation in the News
SalioGen Developing Novel Gene Insertion Therapy for Stargardt Disease
Eye On the Cure Research News-
Apr 12, 2024
Phase 3 Clinical Trial of NAC Launched for RP Patients
Eye On the Cure Research News -
Apr 11, 2024
ViGeneron Launches Clinical Trial of Gene Therapy for RP Caused by CNGA1 Mutations
Eye On the Cure Research News -
Apr 8, 2024
Ocugen to Launch Phase 3 Clinical Trial of Modifier Gene Therapy for RP
Eye On the Cure Research News
Living with vision loss can be difficult, but you don't have to do it alone
View local chaptersGet Involved
-
Take important steps toward a cure by participating in a 5k VisionWalk
Join a Walk -
Join people coming together to find a cure from all over the country
Search Our Events -
Make a difference today and support promising research by donating a one-time gift
Donate Now




